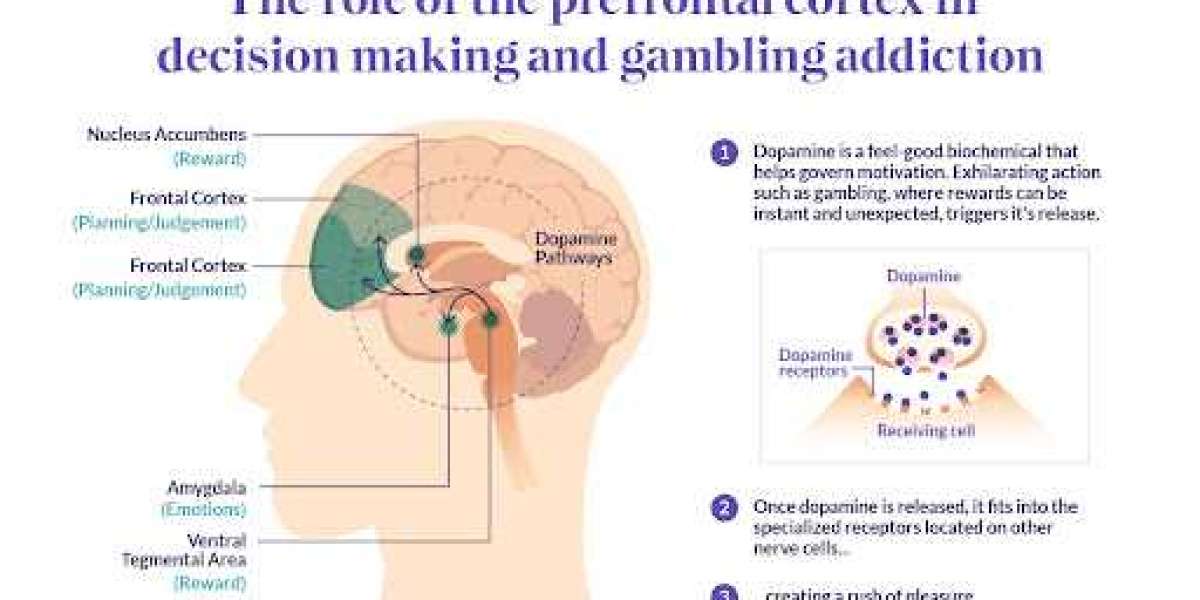
Our density functional theory (DFT) calculations are performed using the Vienna ab initio Simulation Package (VASP). 29 ion-electron interactions are treated by project-enhanced plane wave (PAW) method, and the electron exchange correlation function is treated by Perdew, Burke, and Ernzerhof (PBE). 30 Because the standard PBE functional fails to correctly describe the weak interaction, we use the Grimme VDW-corrected PBE method, adding the semi-empirical dispersion potential to the traditional Kohn-Sham DFT energy. 31 The method introduces the damped atom pair dispersion correction in the form of C6R−6 in the DFT. The accuracy of PBE has been well validated in recent work. 32 The 600 eV cutoff energy of the plane wave base group is used in all calculations, with an energy accuracy of 10−6 eV and a force accuracy of 10− 3 eVA −1. The Monkhorst−Pack k grid centered on 9×9×1Γ is good enough for numerical convergence. A vacuum space of 15A is used in the z direction to prevent interaction effects from adjacent cells.
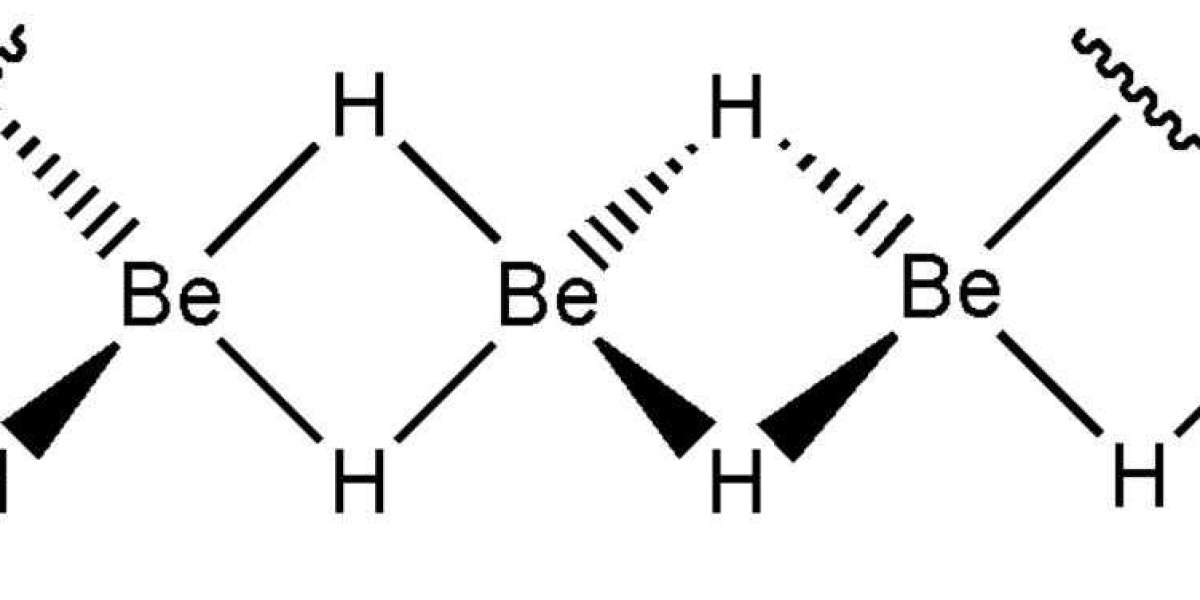
CALYPSO code 33 using particle swarm optimization (PSO) methods is used to search for the lowest energy BeH2 monolayer. The PSO algorithm has been proved to be an effective structure prediction method in many works. 34 The required structural relaxation is performed by the PBE function implemented in the VASP code. In our PSO calculation, the population size is set at 48 and the number of generations remains at 30. In addition to the linear polymer form of BeH2, two low energy stable BeH2 monolayers were obtained. To evaluate the dynamic stability of BeH2 monolayer, phonon dispersion analysis with density functional Perturbation theory (DFPT) was performed using Phonopy code35, 36 implemented in VASP. In the calculation of VASP-DFPT lattice dynamics, a 4×4×1 Q-point grid is used to calculate the interatomic forces. To evaluate thermal stability, ab initio molecular dynamics (AIMD) was performed using the Nose −Hoover algorithm. For accuracy and reliability, a 5×5×1 supercell of Be25H50 was used, with a total simulation time of 20ps.